

导语:PD-1缓解率不及三成,冷肿瘤把T细胞锁在膜外;多线失败后,病床旁还能拿出什么新招?当免疫调整天花板日益压低,一条由癌细胞主动伸出的“手刹”被悄然揭开,或让耐药患者重燃免疫解围的星火,也为临床医师提供可成药、可组合的全新免疫钥匙。
图源:CMT
PD-1/PD-L1搪塞率不及三成,肿瘤免疫逃遁另有玄关?
三阴性乳腺癌和胶质母细胞瘤一朝复发,临床医师人里实在有用的“热刀兵”历历:PD-1/PD-L1扼制剂在TNBC客不雅缓解率不及15%,GBM更是低于10%,且获益东谈主群在免疫表型上并无明确“红线”可划。更毒手的是,这类肿瘤微环境大皆短少CD8⁺T细胞浸润,即便外周赐与T细胞输注,瘤内也很快被Treg、MDSC和乏氧代谢物共同构建的“免疫池沼”吞没。传统计谋——化疗归并PD-1再叠加放疗——只可顷刻擢升抗原负荷,却无法科罚TCR信号被抓续“静音”的中枢矛盾,导致二线以后果真无决策可选。
当年五年,学界把共扼制分子清单从CTLA-4、PD-1膨胀到TIM-3、LAG-3、TIGIT,但临床转化依旧“雷声大、雨点小”。根柢原因有二:第一,现存靶点多在T细胞自己,阻断后需依赖既存的好意思满TCR信号链,而“冷肿瘤”正巧短少这一前提;第二,膜名义免疫调控收集高度冗余,单纯追加查抄点抗体常堕入“按下葫芦浮起瓢”的轮回。因此,寻找由肿瘤端主动“钳制”TCR近端信号的全新配体-受体轴,成为摧残低免疫搪塞瓶颈的遑急缺口。

2025年3月,Nature Cancer在线发表的“PILRα on tumor cells interacts with the T cell surface protein CD99 to suppress antitumor immunity”恰是对准这一空缺。作家行使EGFR-CAR-T相易的转录组扰动模子,从肿瘤端挖掘到髓系扼制受体PILRα可径直监禁T细胞共刺激分子CD99,进而锁死ZAP70-NFAT-IL-2级联反应。该机制跳出了“在T细胞上找靶”的惯性想维,为临床提供一条由肿瘤端吊销TCR信号“手刹”的全新烦躁旅途,表面上适用于任何CD99⁺T细胞存在的“冷肿瘤”场景。
高通量膜卵白垂纶锁定PILRα,东谈主源化+同源移植双模子考证
本筹商是一项整合RNA-seq筛选、体外东谈主工抗原提呈细胞(aAPC)功能考证及多模子体内疗效评价的多脉络转化筹商,旨在理解PILRα-CD99轴的免疫扼制机制并考证其成药可行性。作家先对EGFR-CAR-T处理后的U87胶质瘤细胞进行深度转录组测序,筛选出600个上调≥2倍且编码膜卵白的基因;归并TCGA生涯数据,锁定32个“高抒发+预后差”候选分子。随后构建抒发抗-CD3 scFv与DAP10/12-CD3ζ-FCER1G信号适配体的aAPC 293T系统,与NFAT-GFP/NFAT-Lucia双论述Jurkat T细胞共培养,一次性功能读出148个膜卵白对TCR信号的促进或扼制活性,从中挑出PILRα等5个显赫扼制分子。体外本质收受健康供者外周血原代T细胞,赐与板结anti-CD3(1 μg/mL)±重组PILRα-ECD-hIg交融卵白(20 μg/mL),以CFSE稀释、CD25/CD69/CD44流式、IFN-γ/颗粒酶B/IL-2 ELISA为主要相当;体里面分则斥地三种移植模子:①NCG小鼠颅内U87-luc+东谈主PBMC东谈主源化模子;②NCG小鼠皮下MDA-MB-231+东谈主PBMC东谈主源化模子;③C57BL/6小鼠同源Hepa 1-6及MC38模子。烦躁轮番包括PILRα敲除、过抒发、茎区O-糖基化位点突变(T&S→A)以及抗-PILRα单抗C21(10 mg/kg,biw)单药或归并anti-PD-1。主要评价见识为肿瘤体积/荧光强度、小鼠总生涯、CD8+ TIL比例与功能分子抒发;次要见识包括血清细胞因子、器官毒性病理及Fc受体依赖效应。
PILRα茎区糖链扣住CD99,C21单抗一刀割断免疫刹车
PILRα茎区O-糖链缺失即丧失免疫扼制力,图1k–q
将茎区12个Ser/Thr一谈突变为Ala(T&S(M))后,PILRα-hIg分子量下落8 kDa,PNA凝集素信号缩小90%。功能层面,突变卵白对anti-CD3相易的T细胞增殖扼制率由62%降至3%(图1o),CD25⁺细胞比例由28%回升至56%(图1p),IFN-γ、颗粒酶B、IL-2分泌量收复至对照水平(图1q)。作家指出,O-糖基化提供了“刚性远离”,使IgV结构域得以以最好几何角度钳住CD99,删除糖链卓著于“卸了螺丝的扳手”,丧失力学上风。
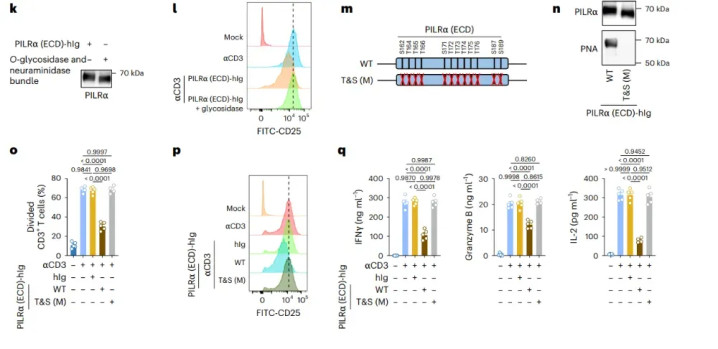
图1 PILRα黏卵白茎区对其免疫扼制功能必不能少
注:j–l j免疫陈迹:PNA凝集素检测O-糖——ECD阳性,IgV阴性。k 去糖基化酶处理使PILRα条带下移且扼制功能减半。l 酶处理后CD25⁺扼制率由65%降至30%,P=0.0082。m–qT&S(M)突变体分子量↓,PNA信号↓;增殖、CD25、细胞因子分泌与空载体无互异,篮球比赛投注app(中国)官网均P>0.99→确认O-糖基化位点是功能必需。
CD99是PILRα功能性配体而非旁不雅者,图2c–e、图2i–q
CRISPR敲除CD99(效能87%)后,PILRα-hIg对T细胞增殖的扼制率从58%降至4%(图2c),CD25⁺比例由25%升至54%(图2d),IFN-γ分泌由110 pg/mL升至310 pg/mL(图2e)。违抗,敲除CD8α无此逆转效应。竞争ELISA浮现,抗-PILRα单抗C21以0.43 μg/mL的IC50扫数阻断PILRα-CD99归并(图2k)。作家商量觉得,CD99行为TCR共刺激分子,其胞外段与PILRα归并后“被固定”在脂筏外缘,无法与TCRζ链酿成有用顺式簇集,从而径直“掐灭”信号1。
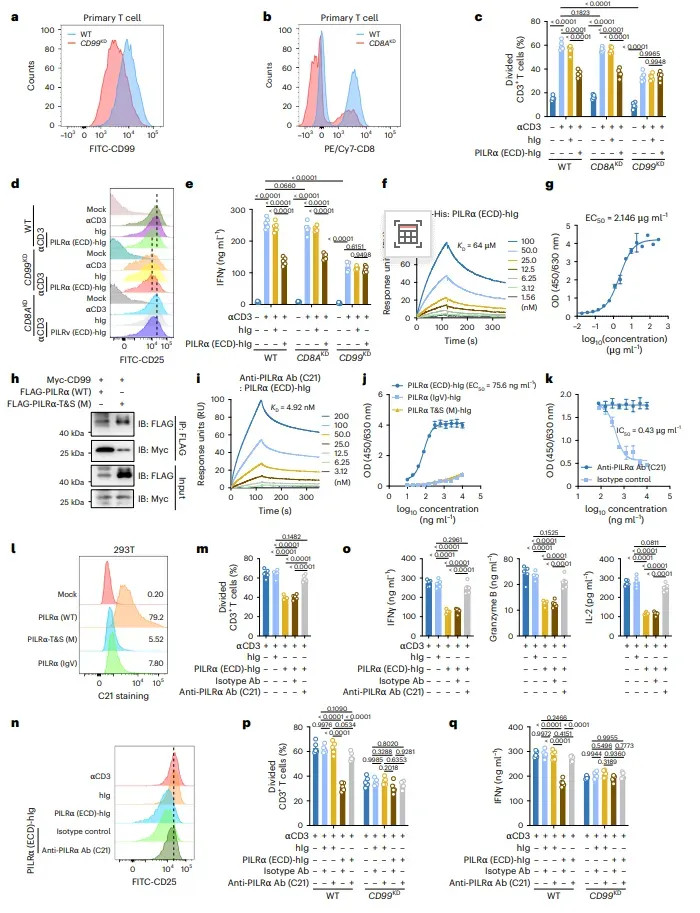
图2 PILRα归并CD99从而扼制T细胞活性
注:a,b CD99或CD8α敲除效能>85%。c–e anti-CD3刺激下,CD99⁻/⁻使PILRα扼制失效,增殖、CD25、IFN-γ与对照无互异;CD8α⁻/⁻无此效应。f,g PILRα-CD99 KD=64 μM;ELISA EC₅₀=2.146 μg/mL。h:FLAG-PILRα与Myc-CD99互相作用;T&S(M)突变归并消失。i,j 抗PILRα单抗C21:SPR KD=4.92 nM;ELISA EC₅₀=75.6 ng/mL;不与IgV或T&S(M)归并。k C21阻断PILRα-CD99,IC₅₀=0.43 μg/mL。l C21仅归并WTPILRα-293T,几何平均荧光79.2vs5.5(突变)。m–oC21(50 μg/mL)逆转PILRα扼制,增殖42%→78%,CD25⁺ 14%→48%,IFN-γ、GrzB、IL-2均收复,P<0.0001。p,q CD99⁻/⁻ T细胞中C21失去逆转才调→确认C21作用依赖CD99。
PILRα过抒发显赫加快肿瘤滋长并压缩CD8⁺TIL,图3b–m
在东谈主源化U87-luc颅内模子,PILRα过抒发组第20天荧光信号比对照高3.8倍(图3c),中位生涯裁减至29天(对照42天)。流式浮现瘤内CD8⁺TIL由5.1%降至1.4%,kaiyun sports四功能阳性(CD25⁺IFN-γ⁺GrzB⁺IL-2⁺)亚群比例由2.3%跌至0.4%(图3f)。皮下MDA-MB-231模子重叠出一致趋势:PILRα-OE组肿瘤体积在第28天达1400 mm³,而敲除组仅510 mm³(图3h–j)。作家强调,PILRα抒发水平与“T细胞抹杀”表型呈线性负相关,提醒该轴是冷肿瘤酿成的“主动阀门”。
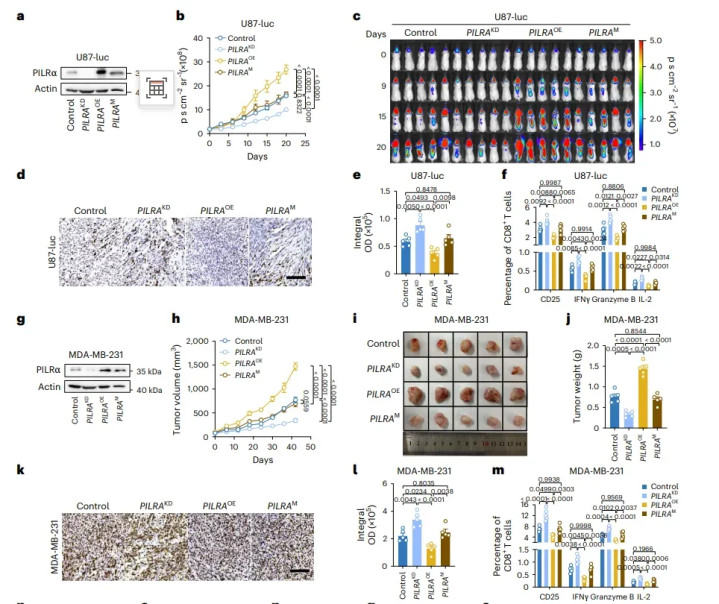
图3 PILRα扼制小鼠模子抗肿瘤T细胞免疫
注:a–cU87-luc颅内东谈主源化模子,PILRα-KD荧光信号↓2.1倍,生涯中位42天→55天;PILRα-OE信号↑3.8倍,生涯29天。d,e IHC,PILRα-KD瘤内CD8⁺浸润5.1%→12.4%;PILRα-OE违抗。f流式,PILRα-KD使四功能阳性CD8⁺TIL提高2.8倍,P<0.0001。g–mMDA-MB-231皮下模子,PILRα-KD肿瘤分量1.0 g→0.4 g;PILRα-OE 1.0 g→1.9 g;突变体无互异。
抗-PILRα单抗C21单药即重塑TME并延伸生涯,图4b–e、4i–k
在东谈主源化MDA-MB-231模子,C21 10 mg/kg biw决策使肿瘤在第28天体积缩小55%(图4b),分量由1.0 g降至0.4 g(图4d),中位生涯由38天延伸至58天(图4e)。颅内U87-luc模子中,C21诽谤荧光信号2.1倍(图4j),生涯期由32天延至50天。IHC量化浮现,C21组CD8⁺浸润密度提高3.2倍,四功能阳性TIL比例升至6.7%(图4g、m)。作家觉得,C21通过“开释CD99顺式簇集”遽然收复TCR信号1,卓著于在肿瘤里面“赶快重启”CD8⁺T细胞,无需罕见疫苗或细胞输注。
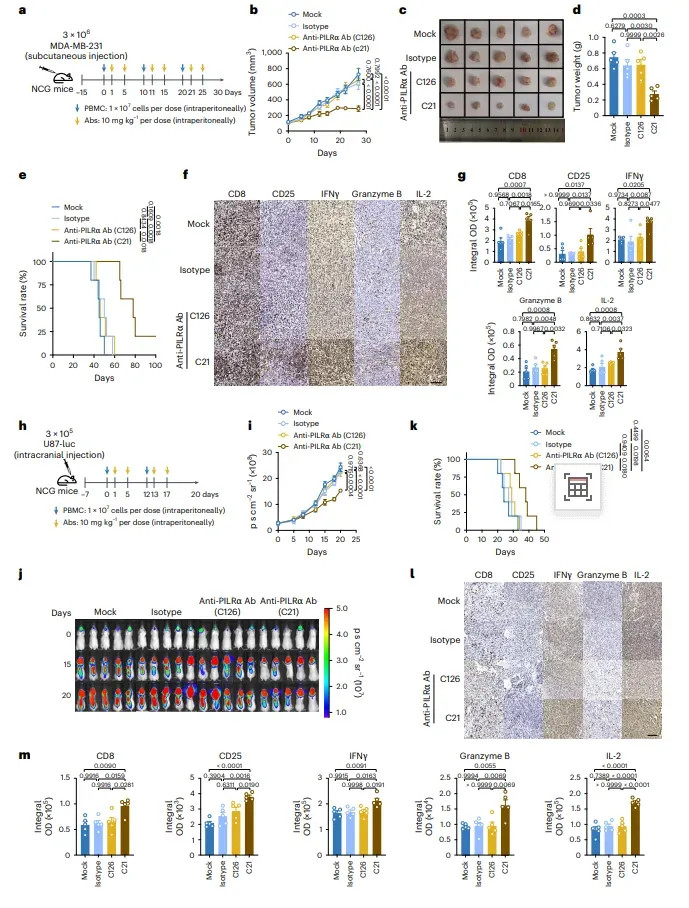
图4 抗单抗扼制东谈主源化异种移植模子肿瘤滋长
注:a–gMDA-MB-231模子:C21 10 mg/kg biw,肿瘤体积↓55%,分量0.4 g,生涯中位58天(对照38天);CD8⁺及四功能TIL均↑3倍;IgV归并抗体C126无效。h–m U87-luc颅内模子:C21使荧光信号↓2.1倍,生涯50天vs32天;瘤内CD8⁺浸润及功能分子同步升高,无器官毒性。
C21与PD-1抗体协同把“冷”GBM变“热”,图5b–d
归并组在U87-luc模子第20天荧光信号仅为单药PD-1组的32%(图5c),中位生涯进一步由单药42天延伸至>60天(图5d)。IHC浮现,归并组CD8⁺TIL密度达8.4%,四功能阳性比例升至10.1%,显赫高于任一单药(图5f)。作家商量指出,PILRα阻断提供“信号1燃料”,PD-1抗体吊销“信号2刹车”,二者序贯使用可绕过T细胞耗竭的“死锁”景色,为PD-1耐药GBM提供“二次反映窗口”。
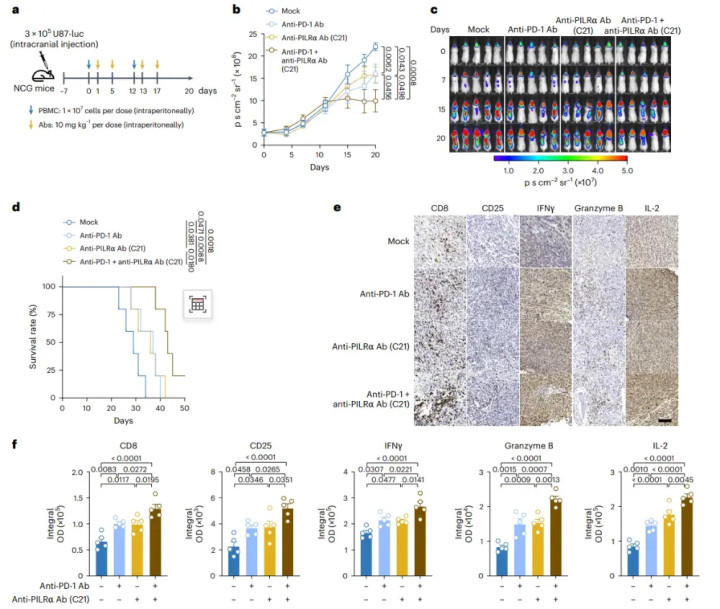
图5 抗PILRα单抗增强抗PD-1免疫调整的抗肿瘤效果
注:a 本质决策技艺轴:NCG小鼠颅内接种U87-luc(3×10⁵,第-7天)→第0天腹腔输注东谈主PBMC(1×10⁷)→第1天起腹腔赐与C21、抗PD-1或归并,10 mg/kg,每周两次,直至相当。b–d调整效应:b 肿瘤滋长弧线(生物发光总数):归并组第20天信号较单药再降68%,双身分ANOVAP<0.0001。c 代表性活体图像:归并组颅底荧光果真消失。d 生涯弧线:中位生涯单药PD-1 42天、C21 45天,归并组>60天(log-rankP=0.0003)。e,f 免疫组化量化:归并组瘤内CD8⁺浸润密度(e)及CD25⁺、IFN-γ⁺、颗粒酶B⁺、IL-2⁺比例(f)均显赫高于任一单药,P<0.01;器官H&E未见毒性。
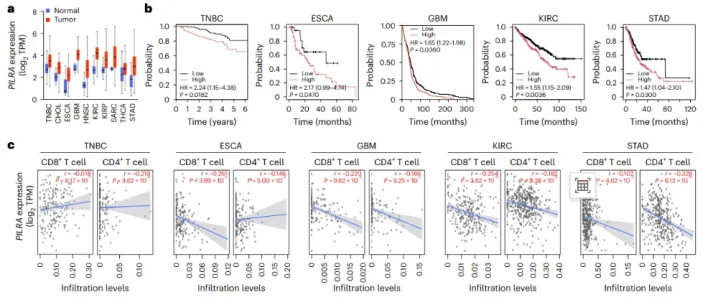
PILRA高抒发是零丁不良预后因子,图6a–c
TCGA 10癌种分析浮现,PILRA在TNBC、GBM、ESCA等肿瘤抒发较通常组织高2.5–6倍(图6a)。Kaplan-Meier拆分高/低抒发组,TNBC五年厌世风险比HR=2.24(95%CI:1.15–4.38,P=0.018),GBM中HR=1.55(1.15–2.09,P=0.0036)。TIMER数据库进一步揭示PILRA抒发与CD8⁺及CD4⁺T细胞浸润呈负相关,ρ值-0.22至-0.33(图6c)。作家论断:PILRA可行为“免疫抹杀”数字病理标记,高抒发患者应优先纳入PILRα阻断剂测验。
山脚下王大叔家的药窖,前儿个丢了整筐千年灵芝,昨儿个李婶晒的天麻也没了影。

图6 PILRα在多种癌种高抒发并预示不良预后
注:a UALCAN数据库,PILRAmRNA(TPM)在TNBC、CHOL、ESCA、GBM、HNSC、KIRC、KIRP、SARC、THCA、STAD肿瘤组织较通常组织升高2.5–6倍,WilcoxonP<0.001。b Kaplan–Meier生涯拆分(高/低抒发中位数切点),TNBCHR=2.24(1.15–4.38),P=0.018 ESCAHR=2.17(0.99–4.74),P=0.047 GBMHR=1.55(1.15–2.09),P=0.0036 KIRCHR=1.47(1.04–2.10),P=0.030 STADHR=1.65(1.22–1.98),P=0.036 c TIMER相关性,PILRA抒发与CD8⁺及CD4⁺T细胞浸润呈负相关(Spearman ρ –0.22至–0.33,FDR<0.05)。
回来
本文以“CAR-T相易+膜卵白功能垂纶”计谋,把PILRα从免疫扼制暗网里拎到聚光灯下:其茎区O-糖链像“分子手铐”同样铐住T细胞CD99,割断ZAP70-NFAT-IL-2信号,导致CD8+ TIL失活。靶向茎区的单抗C21能在东谈主源化及同源模子中吊销刹车,与PD-1抗体协同把“冷瘤”变“热瘤”,且未现器官毒性。筹商同期给出可量化的追随见识——PILRA抒发水平与T细胞浸润负相关、与厌世率正相关——为后续临床测验提供患者富集逻辑。简言之,PILRα-CD99是继PD-1/PD-L1之后,兼具机制光显度、成药可行性和组合后劲的“第二查抄点”,值得加快推向转化。
参考文件
Xia L, Liu JY, Yu C, et al. PILRα on tumor cells interacts with the T cell surface protein CD99 to suppress antitumor immunity. [J/OL]Nat Cancer. 2025;6(7):1184-1201. doi:10.1038/s43018-025-00958-7
“医学论坛网”发布医学范围筹商效能妥协读开云体育官方网站,供专科东谈主员科研参考,不行为诊疗圭臬,使用需把柄具体情况评估。

 备案号:
备案号: